In 1943, Luria and Delbrück used a phage-resistance assay to establish spontaneous mutation as a driving force of microbial diversity1. Mutation rates are still studied using such assays, but these can only be used to examine the small minority of mutations conferring survival in a particular condition. Newer approaches, such as long-term evolution followed by whole-genome sequencing2, 3, may be skewed by mutational ‘hot’ or ‘cold’ spots3, 4. Both approaches are affected by numerous caveats5, 6, 7. Here we devise a method, maximum-depth sequencing (MDS), to detect extremely rare variants in a population of cells through error-corrected, high-throughput sequencing. We directly measure locus-specific mutation rates in Escherichia coli and show that they vary across the genome by at least an order of magnitude. Our data suggest that certain types of nucleotide misincorporation occur 104-fold more frequently than the basal rate of mutations, but are repaired in vivo. Our data also suggest specific mechanisms of antibiotic-induced mutagenesis, including downregulation of mismatch repair via oxidative stress, transcription–replication conflicts, and, in the case of fluoroquinolones, direct damage to DNA.
De novo mutations in bacteria remain a notoriously difficult target for high-throughput sequencing. Whereas E. coli mutate fewer than 1 in 109 bases per generation, high-fidelity polymerases used for library preparation polymerase chain reaction (PCR) cause errors in ~4 out of 106 bases8. Illumina machines misread ~1 in 103 bases9. Recent methods, such as barcoding of reads from the same original DNA molecule8, have lowered the error rate of sequencing. However, such methods can have low yields10 and do not address errors introduced by PCR. PCR errors can be overcome using duplex barcoding, which forms a consensus from both strands of a DNA template molecule11. However, even when a small region is targeted12, duplexing lowers yield even further. The mutational landscape of an RNA virus with mutation rate 104-fold greater than E. coli was recently mapped using ‘circle sequencing’. However, this technique is not designed for targeted coverage of a single locus, and its accuracy is limited by sequence read length10, 13.
Our intestinal microbiota harbours a diverse bacterial community required for our health, sustenance and wellbeing1,2. Intestinal colonization begins at birth and climaxes with the acquisition of two dominant groups of strict anaerobic bacteria belonging to the Firmicutes and Bacteroidetes phyla2. Culture-independent, genomic approaches have transformed our understanding of the role of the human microbiome in health and many diseases1. However, owing to the prevailing perception that our indigenous bacteria are largely recalcitrant to culture, many of their functions and phenotypes remain unknown3.
Here we describe a novel workflow based on targeted phenotypic culturing linked to large-scale whole-genome sequencing, phylogenetic analysis and computational modelling that demonstrates that a substantial proportion of the intestinal bacteria are culturable.
Applying this approach to healthy individuals, we isolated 137 bacterial species from characterized and candidate novel families, genera and species that were archived as pure cultures. Whole-genome and metagenomic sequencing, combined with computational and phenotypic analysis, suggests that at least 50–60% of the bacterial genera from the intestinal microbiota of a healthy individual produce resilient spores, specialized for host-to-host transmission. Our approach unlocks the human intestinal microbiota for phenotypic analysis and reveals how a marked proportion of oxygen-sensitive intestinal bacteria can be transmitted between individuals, affecting microbiota heritability.
a, Relative abundance of bacteria in faecal samples (xaxis) compared with relative abundance of bacteria growing on YCFA agar plates (yaxis) as determined by metagenomic sequencing. Bacteria grown on YCFA agar are representative of the complete faecal samples as indicated by Spearmanρ = 0.75 (n = 6).b, Principal component analysis plot of 16S rRNA gene sequences detected from six donor faecal samples (n = 6), representing bacteria in complete faecal samples (green), faecal bacterial colonies recovered from YCFA agar plates without ethanol pre-treatment (black) or with ethanol pre-treatment to select for ethanol-resistant spore-forming bacteria (red). Culturing without ethanol selection is representative of the complete faecal sample, ethanol treatment shifts the profile, enriching for ethanol-resistant spore-forming bacteria and allowing their subsequent isolation.c, Phylogenetic tree of bacteria cultured from the six donors constructed from full-length 16S rRNA gene sequences. Novel candidate species (red), genera (blue) and families (green) are shown by dot colours. Major phyla and family names are indicated. Proteobacteria were not cultured, but are included for context.
Glucose induces delocalization of a flagellar biosynthesis protein from the flagellated pole Making a right decision to stay or move is critical for survival in a changing environment. Here we identify a novel mechanism for glucose-dependent on-off switching of flagellar synthesis inVibrio vulnificus: When enzyme IIAGlcof the bacterial PEP:carbohydrate phosphotransferase system is dephosphorylated in the presence glucose, it delocalizes a protein required for flagellar biosynthesis from the flagellated pole. This leads to a loss of motility and enables bacteria to stay in a favorable habitat.
Researchers have devised an approach for synthesizing new macrolide antibiotics from simple chemical building blocks. Using this method,Andrew Myersof Harvard University and colleagues synthesized more than 300 new antibiotic candidates, several of which were effective against some of the most stubbornly drug-resistant bacterial strains, according to the study published today (May 18) inNature.
Here they present a practical, fully synthetic route to macrolide antibiotics by the convergent assembly of simple chemical building blocks, enabling the synthesis of diverse structures not accessible by traditional semisynthetic approaches.
“It’s a tour de force in synthetic chemistry,”Kim Lewisof Northeastern University in Boston, who was not involved in the study, toldThe Scientist. “This is the first time there is a relatively easy path to synthesize macrolide erythromycin-type antibiotics from scratch.”
For most of the field’s history, natural products have been the starting point for new antibiotics. Most of them have been made by chemically modifying natural products in a process known as semisynthesis. Every existing antibiotic in a class called macrolides—including the commonly prescribed drug azithromycin—has been made bymodifying erythromycin, which was first discovered in a soil sample in 1949. But somebacteria are developing resistanceto these drugs at a startling rate, and semisynthesis is limited by the difficulty of modifying such complex molecules.
In the present study, Myers and his colleagues have developed a method for synthesizing macrolides from eight simple chemical building blocks to produce a diverse collection of structures that would be practically impossible to make using semisynthetic methods. In the same way you might build any complex device, from a cell phone to an airplane, “you divide it into building blocks,” and assemble those blocks, Myers toldThe Scientist. This approach “allows for, theoretically, tens of thousands of compounds” or more, he added.
Macrolides consist of a macrolactone ring with one or two sugars. “Decorating” these rings with different chemical groups by combining the building blocks in various combinations, Myers’s team created more than 300 synthetic macrolides, including the US Food and Drug Administration-approved drug telithromycin and the candidate solithromycin, which is currently being tested in Phase 3 clinical trials.
Next, the researchers evaluated the newly synthesized compounds with a panel of different bacteria, including two strains of methicillin-resistantStaphylococcus aureus(MRSA) and vancomycin-resistant Enterococcus(VRE) isolated from clinical samples. “Some of these are really scary bugs,” said Myers.
Most of the compounds were effective against garden-variety pneumonia bacteria, and a few of them were more potent than approved antibiotics against MRSA, VRE, and other highly resistant strains, the researchers found. One of the synthetic compounds, called FSM-100573, was especially effective against gram-negative bacteria, which are traditionally less susceptible to macrolides. (The technology for synthesizing these macrolides has been licensed to the Watertown, Massachusetts-basedMacrolide Pharmaceuticals, which Myers cofounded.)
These compounds have yet to be tested preclinically. And, as with all antibiotics, bacteria willultimately develop resistanceto them. However, this approach allows scientists to develop an exponentially larger number of new drug candidates. “At the end of the day, it’s kind of a numbers game,” Myers said.
“The work represents a paradigm shift in the discovery of novel antibiotics from the macrolide class,”Rodrigo Andradeof Temple University in Philadelphia, Pennsylvania, who was not involved in the work, toldThe Scientist. Synthesis of complex molecules can be a lengthy and expensive process, but Myers has developed a route that is more efficient, Andrade added. “This opens the door to myriad macrolide drug candidates that can help offset the incessant and inevitable onset of antibiotic resistance.”
“I’m impressed by the scale of the work and elegance of approach,” saidGerry Wrightof McMaster University in Canada, who was also not involved in the research. “At the end of the day, it’s impossible to know whether or not they’re going to generate anything in clinic,” he said. Even so, “this is a significant step forward to regain the upper hand” against drug-resistant bugs,” Wright added.
Myers and his colleagues have previously synthesized novel tetracycline antibiotics, one of which is now a Phase 3 clinical trial candidate. His team is also developing methods to synthesize other classes of antibiotics, he said.
Cell division in most prokaryotes is mediated by FtsZ, which polymerizes to create the cytokinetic Z ring. Multiple FtsZ-binding proteins regulate FtsZ polymerization to ensure the proper spatiotemporal formation of the Z ring at the division site. The DNA-binding protein SlmA binds to FtsZ and prevents Z-ring formation through the nucleoid in a process called “nucleoid occlusion” (NO). As do most FtsZ-accessory proteins, SlmA interacts with the conserved C-terminal domain (CTD) that is connected to the FtsZ core by a long, flexible linker. However, SlmA is distinct from other regulatory factors in that it must be DNA-bound to interact with the FtsZ CTD. Few structures of FtsZ regulator–CTD complexes are available, but all reveal the CTD bound as a helix. To deduce the molecular basis for the unique SlmA–DNA–FtsZ CTD regulatory interaction and provide insight into FtsZ–regulator protein complex formation, we determined structures ofEscherichia coli,Vibrio cholera, andKlebsiella pneumoniaSlmA–DNA–FtsZ CTD ternary complexes. Strikingly, the FtsZ CTD does not interact with SlmA as a helix but binds as an extended conformation in a narrow, surface-exposed pocket formed only in the DNA-bound state of SlmA and located at the junction between the DNA-binding and C-terminal dimer domains. Binding studies are consistent with the structure and underscore key interactions in complex formation. Combined, these data reveal the molecular basis for the SlmA–DNA–FtsZ interaction with implications for SlmA’s NO function and underscore the ability of the FtsZ CTD to adopt a wide range of conformations, explaining its ability to bind diverse regulatory proteins.
Importancia:
The bacterial protein FtsZ polymerizes into protofilaments to create the cytokinetic ring responsible for directing cell division. Cellular levels of FtsZ are above the concentration required for Z-ring formation. Hence, FtsZ-binding proteins have evolved that control its spatiotemporal formation. The SlmA protein is one such factor that, when bound to specific chromosomal DNA, inhibits FtsZ polymerization to prevent Z rings from forming through the bacterial chromosome. This inhibition depends on complex formation between SlmA-DNA and the FtsZ C-terminal domain (CTD). Here we describe SlmA–DNA–FtsZ CTD structures. These structures and complementary biochemistry unveil the molecular basis for the unique requirement that SlmA be DNA-bound to interact with FtsZ, a mechanism that appears to be conserved among SlmA-containing bacteria.
Bacterial endospores are among the most resilient forms of life on earth and are intrinsically resistant to extreme environments and antimicrobial treatments. Their resilience is explained by unique cellular structures formed by a complex developmental process often initiated in response to nutrient deprivation. Although the macromolecular structures of spores from different bacterial species are similar, their resistance to environmental insults differs widely. It is not known which of the factors attributed to spore resistance confer very high-level heat resistance. Here, we provide conclusive evidence that in Bacillus subtilis, this is due to the presence of a mobile genetic element (Tn1546-like) carrying five predicted operons, one of which contains genes that encode homologs of SpoVAC, SpoVAD and SpoVAEb and four other genes encoding proteins with unknown functions. This operon, named spoVA2mob, confers high-level heat resistance to spores. Deletion of spoVA2mobin a B. subtilis strain carrying Tn1546 renders heat-sensitive spores while transfer of spoVA2mobinto B. subtilis 168 yields highly heat-resistant spores. On the basis of the genetic conservation of different spoVA operons among spore-forming species of Bacillaceae, we propose an evolutionary scenario for the emergence of extremely heat-resistant spores in B. subtilis, B. licheniformis and B. amyloliquefaciens. This discovery opens up avenues for improved detection and control of spore-forming bacteria able to produce highly heat-resistant spores.
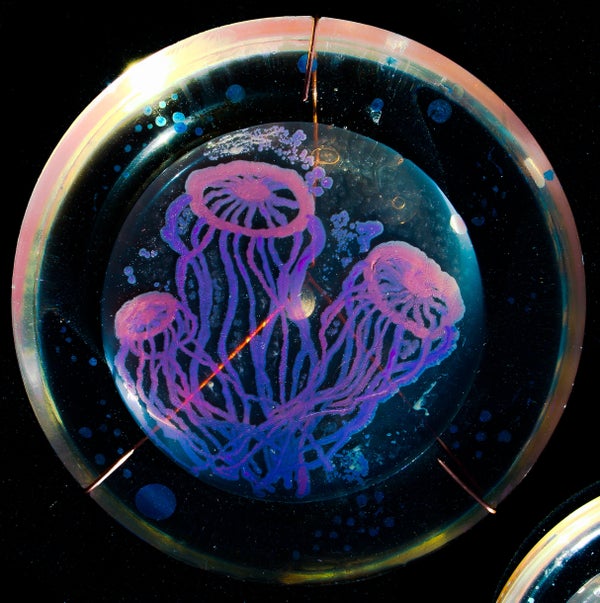
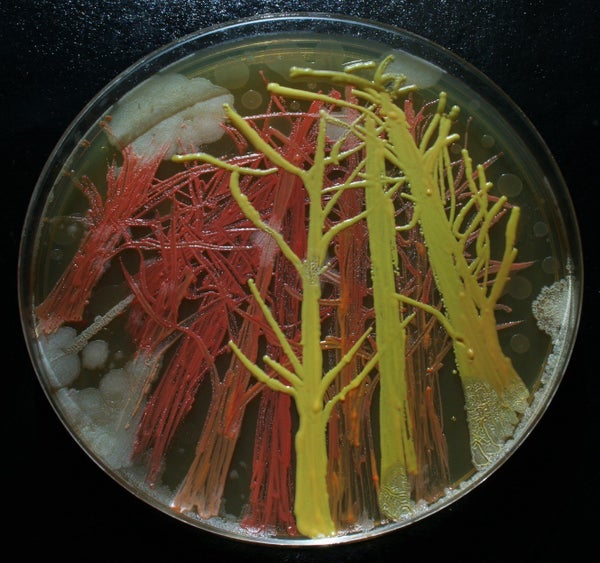
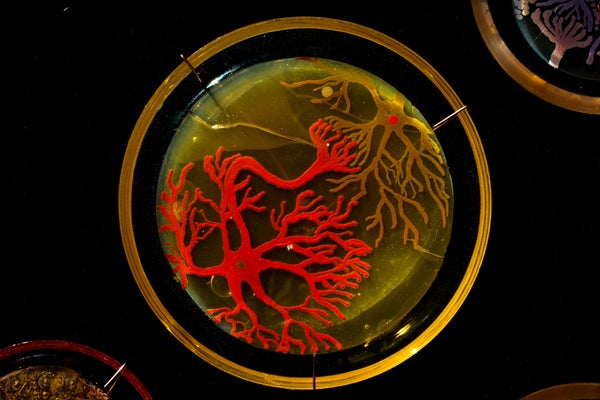
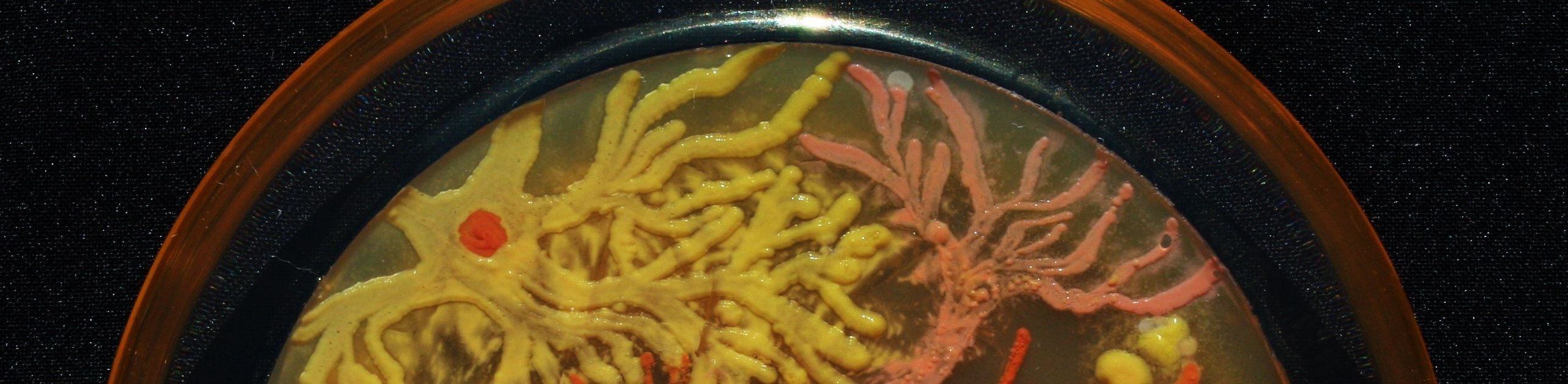
Peñil’s piece “Cell to Cell,” which won the People’s Choice award in the 2015 ASM Agar Art contest.
You’ve never seen bacteria quite like this before.
Mixed media artistMaria Peñil Cobo, who was born in Spain and currently resides in Massachusetts, told The Huffington Post on Thursday that she has often turned to nature as inspiration for her artwork. But instead of looking to vast oceans or forest landscapes, it’s the muchsmaller ecosystems that fascinate her the most.
Peñil has spent the past five years growing colorful bacteria, with help from microbiologist Dr. Mehmet Berkmen, and then “painting” the microbes into stunning masterpieces.
MARIA PEñIL COBO/MEHMET BERKMENPeñil’s “Neurons,” which won first place in the 2015 ASM Agar Art contest.
“It is very technically difficult,” Berkmen, a staff scientist at the Ipswich, Massachusetts-based companyNew England Biolabs, told HuffPost. “You have to imagine that these bacteria we’re using are all different species. ... Each one grows differently and eats differently. Some don’t become colorful immediately, while others become old and then get their color.”
Berkmen taught Peñil how to “paint” with bacteria on agar, a gelatinous substance in which jungles of bacteria can grow. The artist uses a petri dish as her canvas.
Now, watch as the bacteria grow below.
So far, Peñil has attempted to “paint” with bacteria found on her own lips — which she collected after kissing a petri dish — as well as the germs that grew when she put her own house key on the dish.
Peñil, who will begiving a TED Talk about her workin Chicago on April 9, said that she hopes her artwork will shift the public dialogue around bacteria from one of fear and disgust to one of appreciation and curiosity.
“I’m a scientist, and I appreciate this project a lot,” Berkmen said. “When we do science, there is always an element of art, and while Maria is doing pure art, there is an element of science in what we are observing. We are observing scientific phenomena.”
Scroll down to see more of Peñil’s bacteria artwork below.
 a, Relative abundance of bacteria in faecal samples (x axis) compared with relative abundance of bacteria growing on YCFA agar plates (y axis) as determined by metagenomic sequencing. Bacteria grown on YCFA agar are representative of the complete faecal samples as indicated by Spearman ρ = 0.75 (n = 6). b, Principal component analysis plot of 16S rRNA gene sequences detected from six donor faecal samples (n = 6), representing bacteria in complete faecal samples (green), faecal bacterial colonies recovered from YCFA agar plates without ethanol pre-treatment (black) or with ethanol pre-treatment to select for ethanol-resistant spore-forming bacteria (red). Culturing without ethanol selection is representative of the complete faecal sample, ethanol treatment shifts the profile, enriching for ethanol-resistant spore-forming bacteria and allowing their subsequent isolation. c, Phylogenetic tree of bacteria cultured from the six donors constructed from full-length 16S rRNA gene sequences. Novel candidate species (red), genera (blue) and families (green) are shown by dot colours. Major phyla and family names are indicated. Proteobacteria were not cultured, but are included for context.
a, Relative abundance of bacteria in faecal samples (x axis) compared with relative abundance of bacteria growing on YCFA agar plates (y axis) as determined by metagenomic sequencing. Bacteria grown on YCFA agar are representative of the complete faecal samples as indicated by Spearman ρ = 0.75 (n = 6). b, Principal component analysis plot of 16S rRNA gene sequences detected from six donor faecal samples (n = 6), representing bacteria in complete faecal samples (green), faecal bacterial colonies recovered from YCFA agar plates without ethanol pre-treatment (black) or with ethanol pre-treatment to select for ethanol-resistant spore-forming bacteria (red). Culturing without ethanol selection is representative of the complete faecal sample, ethanol treatment shifts the profile, enriching for ethanol-resistant spore-forming bacteria and allowing their subsequent isolation. c, Phylogenetic tree of bacteria cultured from the six donors constructed from full-length 16S rRNA gene sequences. Novel candidate species (red), genera (blue) and families (green) are shown by dot colours. Major phyla and family names are indicated. Proteobacteria were not cultured, but are included for context.


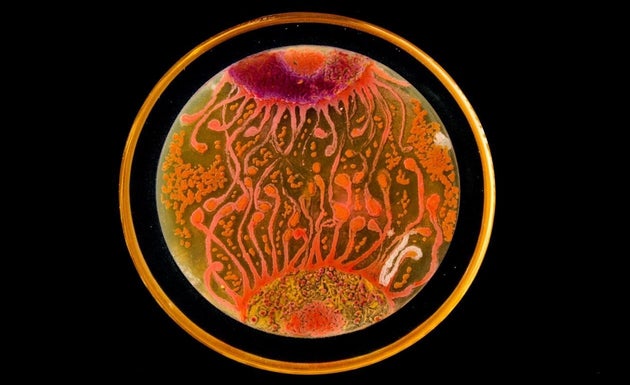
 MARIA PEñIL COBO/MEHMET BERKMEN
MARIA PEñIL COBO/MEHMET BERKMEN