Cyanobacteria were responsible for the origin of oxygenic photosynthesis, and have since come to colonize almost every environment on Earth. Here we show that their ecological range is not limited by the presence of sunlight, but also extends down to the deep terrestrial biosphere. We report the presence of microbial communities dominated by cyanobacteria in the continental subsurface using microscopy, metagenomics, and antibody microarrays. These cyanobacteria were related to surface rock-dwelling lineages known for their high tolerance to environmental and nutritional stress. We discuss how these adaptations allow cyanobacteria to thrive in the dark underground, a lifestyle that might trace back to their nonphotosynthetic ancestors.
Here we present molecular, microscopic, and metagenomic evidence that cyanobacteria predominate in deep subsurface rock samples from the Iberian Pyrite Belt Mars analog (southwestern Spain). Metagenomics showed the potential for a hydrogen-based lithoautotrophic cyanobacterial metabolism. Collectively, our results suggest that they may play an important role as primary producers within the deep-Earth biosphere. Our description of this previously unknown ecological niche for cyanobacteria paves the way for models on their origin and evolution, as well as on their potential presence in current or primitive biospheres in other planetary bodies, and on the extant, primitive, and putative extraterrestrial biospheres.
Distribution of cyanobacteria and other microbial taxa in core samples from the Iberian Pyrite Belt deep subsurface. (A) Relative abundance of different cyanobacterial clades based on 16S rRNA amplicons (bars) and hydrogen concentrations (purple area) across the borehole samples. Hydrogen concentrations are shown relative to the sample with the highest hydrogen concentration. (B) Taxonomic composition of the metagenomic reads from samples 420 and 607.
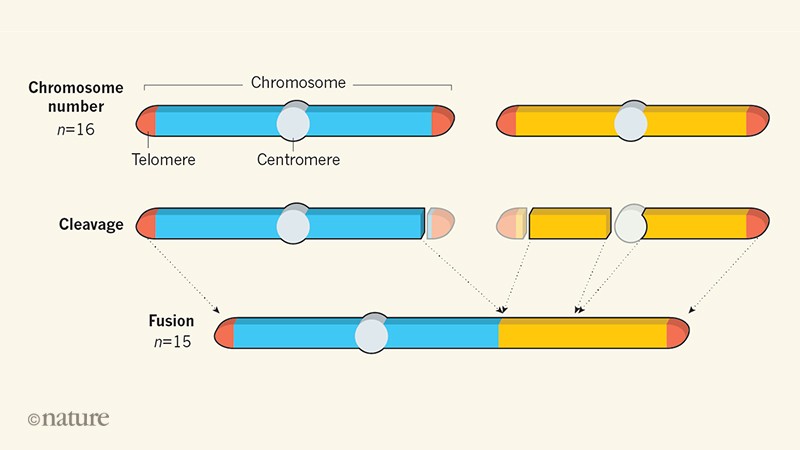
Extant species have wildly different numbers of chromosomes, even among taxa with relatively similar genome sizes (for example, insects)1,2. This is likely to reflect accidents of genome history, such as telomere–telomere fusions and genome duplication events3,4,5. Humans have 23 pairs of chromosomes, whereas other apes have 24. One human chromosome is a fusion product of the ancestral state6. This raises the question: how well can species tolerate a change in chromosome numbers without substantial changes to genome content? Many tools are used in chromosome engineering in Saccharomyces cerevisiae7,8,9,10, but CRISPR–Cas9-mediated genome editing facilitates the most aggressive engineering strategies. Here we successfully fused yeast chromosomes using CRISPR–Cas9, generating a near-isogenic series of strains with progressively fewer chromosomes ranging from sixteen to two. A strain carrying only two chromosomes of about six megabases each exhibited modest transcriptomic changes and grew without major defects. When we crossed a sixteen-chromosome strain with strains with fewer chromosomes, we noted two trends. As the number of chromosomes dropped below sixteen, spore viability decreased markedly, reaching less than 10% for twelve chromosomes. As the number of chromosomes decreased further, yeast sporulation was arrested: a cross between a sixteen-chromosome strain and an eight-chromosome strain showed greatly reduced full tetrad formation and less than 1% sporulation, from which no viable spores could be recovered. However, homotypic crosses between pairs of strains with eight, four or two chromosomes produced excellent sporulation and spore viability. These results indicate that eight chromosome–chromosome fusion events suffice to isolate strains reproductively. Overall, budding yeast tolerates a reduction in chromosome number unexpectedly well, providing a striking example of the robustness of genomes to change.
Bertrand Routy earned a lamentable reputation with Parisian oncologists in 2015. A doctoral student at the nearby Gustave Roussy cancer centre, Routy had to go from hospital to hospital collecting stool samples from people who had undergone cancer treatments. The doctors were merciless. “They made fun of me,” Routy says. “My nickname was Mr Caca.”
But the taunting stopped after Routy and his colleagues published evidence that certain gut bacteria seem to boost people’s response to treatment1. Now, those physicians are eager to analyse faecal samples from their patients in the hope of predicting who is likely to respond to anticancer drugs. “It was an eye-opener for a lot of people who couldn’t see the clinical relevance of gut microbes,” says Routy, who is now at the University of Montreal Health Centre in Canada.
Cancer has been a late bloomer in the microbiome revolution that has surged through biomedicine. Over the past few decades, scientists have linked the gut’s composition of microbes to dozens of seemingly unrelated conditions — from depression to obesity. Cancer has some provocative connections as well: inflammation is a contributing factor to some tumours and a few types of cancer have infectious origins. But with the explosive growth of a new class of drug — cancer immunotherapies — scientists have been taking a closer look at how the gut microbiome might interact with treatment and how these interactions might be harnessed.
After preliminary findings in mice and humans revealed that gut bacteria can sway responses to such drugs, scientists started trying to decipher the mechanisms involved. And researchers are launching a handful of clinical trials that will test whether the gut microbiome can be manipulated to improve outcomes.
Some proponents say that strategies to mould the microbiome could be game-changing in cancer treatment. “It’s a smart place to be,” says Jennifer Wargo, a surgeon–scientist at MD Anderson Cancer Center in Houston, Texas. But others are worried that the move to the clinic is premature. William Hanage, an epidemiologist at the Harvard T. H. Chan School of Public Health in Boston, Massachusetts, calls the idea “phenomenally interesting”, but adds: “I have some anxiety about the notion that only beneficial effects are possible.”
Intriguing link
Although the excitement about microbes and immunotherapy has emerged only in the past three years, some researchers have been exploring connections between gut bacteria and cancer for much longer. Scientists first linked the infectious bacterium Helicobacter pylorito gastric cancer back in the 1990s, for example. And since then, other bacteria have been associated with cancer initiation and progression. Some of these microbes activate inflammatory responses and disrupt the mucus layers that protect the body from outside invaders, creating an environment that supports tumour growth. In other cases, they promote cancer survival by making cells resistant to anticancer drugs.
But gut bacteria can also help fight tumours. In 2013, a group led by Laurence Zitvogel2 at Gustave Roussy and one led by immunologists Romina Goldszmid and Giorgio Trinchieri3 at the National Cancer Institute in Bethesda, Maryland, showed that some cancer treatments rely on the gut microbiome activating the immune system.
Zitvogel’s team found that the chemotherapy drug cyclophosphamide damages the mucus layer that lines the intestine, allowing some gut bacteria to travel into the lymph nodes and spleen, where they activate specific immune cells. For mice raised without microbes in their guts or given antibiotics, the drug largely lost its anticancer effects.
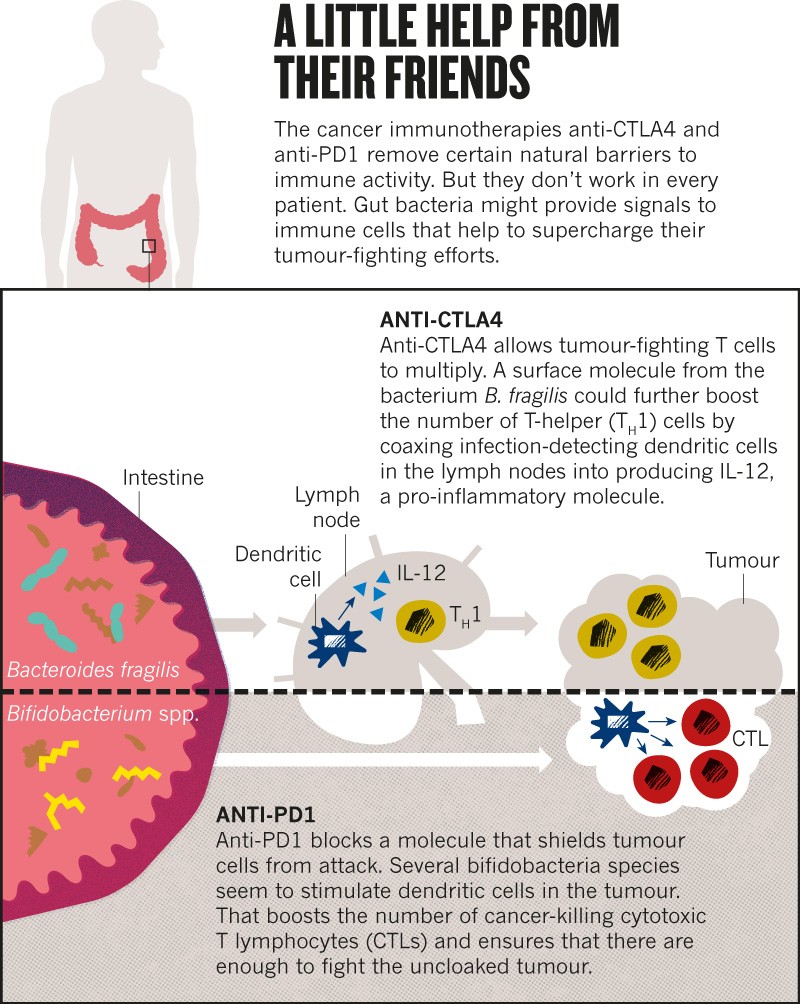
Following this observation, Zitvogel decided to explore whether bacteria in the gut might influence responses to a class of immunotherapy drugs called checkpoint inhibitors. These drugs, typically antibodies to cell-surface molecules such as CTLA4 and PD1, unleash a person’s immune system against tumour cells, and are used to treat several types of cancer (see ‘A little help from their friends’). But only 20–40% of people respond to treatment4.
In 2015, Zitvogel and her team showed that microbe-free mice failed to respond to one such drug, and mice given a particular bacterium, Bacteroides fragilis, responded better than did mice without it5.
The idea started to spread. Thomas Gajewski, a cancer clinician at the University of Chicago in Illinois, reported that Bifidobacterium microbes increased the response to cancer immunotherapy in mice6. These gut-dwelling bacteria acted by boosting the ability of some immune cells to initiate a response against tumours.
Wargo saw these results presented at a meeting in 2014, and on returning to Texas, immediately started to collect stool samples from people with skin cancer who were about to undergo immunotherapy at her institution. Last November, Wargo7, Gajewski8 and Zitvogel1 all published results in Science linking positive immunotherapy responses in people to specific varieties of gut bacteria. The samples that Routy had collected in Paris helped Zitvogel’s team to also show that people who had taken antibiotics for unrelated infections tended to respond poorly to immunotherapy.
To solidify the relationships, the researchers transferred bacteria from the human participants into the intestines of mice with comparable cancers. Rodents who got ‘beneficial’ bacteria developed smaller tumours than did mice that received microbes from people who hadn’t responded to treatment. “All of this work has been very exciting,” says Neeraj Surana, a microbiologist at Boston Children’s Hospital. “They’ve opened up the possibility for a clear therapeutic application of microbiome science.”
Heading to the clinic
Researchers are now running with that possibility. Hassane Zarour, an immunologist at the University of Pittsburgh in Pennsylvania, partnered with the global pharmaceutical company Merck to collect faecal bacteria from people who respond to treatment with a checkpoint inhibitor and transfer them into the intestine of non-responders, a process called faecal microbiome transplant. Merck has invested about US$900,000 into this trial, which is set to start in the next few weeks.
Wargo is planning a similar trial. Together with the Parker Institute for Cancer Immunotherapy in San Francisco, California, and the biotech company Seres Therapeutics in Cambridge, Massachusetts, she expects to test whether faecal transplants can reshape the gut microbiome of non-responders in a beneficial way.
These microbiome transplants are becoming a mainstream treatment for some non-cancer illnesses. In February, for example, the Infectious Diseases Society of America recommended that physicians use these procedures to treat people with bowel infections caused by the bacterium Clostridium difficile who had failed to respond to other treatments. But the approach has downsides. To avoid the risk of inadvertently infecting people with pathogenic microbes, researchers must be careful in how they select donors and screen faecal material before transferring it to recipients. That’s why, in addition to faecal transplants, Seres Therapeutics, the Parker Institute and Wargo will test a pill containing a set of spore-forming bacteria that have been purified from the faeces of responding patients.
Gajewski and his partners at Evelo Biosciences, a biotech company in Cambridge, are using a similar approach. Their trial will assess the effects of two pills containing single bacterial strains in people with different types of cancer, including colon and skin cancer.
Zitvogel is not planning to start clinical trials but she has co-founded the Delaware-based start-up EverImmune, which is developing a microbiome-based pill.
It’s still unclear exactly how microbes might interact with immunotherapeutics. A widely accepted hypothesis is that some boost the body’s response against tumours by regulating how easy it is to activate the immune system. But the precise mechanisms, including which bacteria modulate which immune cells, remain a mystery.
The researchers hope that the clinical trials will help to clarify things. Wargo, for instance, is exploring bacterial metabolites. Her team hopes to find specific metabolic signatures of a good outcome in the stools and blood of people who respond to therapy, as well as to document the numbers of immune cells in the blood and tumours of trial participants.
Gajewski suggests that microbes might be unleashing the immune response by stimulating the gut cells to produce certain molecules. His team is testing whether circulating immune-cell precursors change their behaviour when specific bacteria are given to mice. At the same time, the group is trying to pin down which species might be driving the positive outcomes.
Too early, or just right?
Given the uncertainties, some scientists argue that testing these approaches in humans is risky. Some trial participants could experience side effects, Surana says. And changing the make-up of an individual’s microbiome might predispose them to other health problems.
Faecal transplants come with a lot of unknowns. They have proved safe and effective in many people without cancer, Wargo says, but they have also been associated with unexpected effects, including one case in which the procedure led to weight gain and obesity9. “Should we look for safety signals on these trials? Absolutely.” Wargo says, “But I strongly feel that we need to go into these trials. We need to design them well. We need to really learn from these trials.”
Gajewski, who plans to test the effects of just one bifidobacterial strain at a time, says there’s good reason to be confident. “People have eaten bifidobacteria for a thousand years,” he says. The bacteria are present in the gut of infants and decline in number as the people grow up, so they should at least be safe, he adds.
But it’s unclear whether a single species can help people with cancer and, if so, what bacterium that is. The papers published in Science last year all associated different bacteria with the best outcomes, even for the same cancer and therapy.
The researchers looked at people with cancer from France and the United States, so diet could account for some of the differences, Wargo says. But variations in sample collection, data analysis and statistical methods could also have skewed the results, says Joël Doré, a biologist at the French National Institute for Agricultural Research in Paris who in 2011 helped to launch the International Human Microbiome Standards (IHMS) project with the aim of improving data reproducibility in microbiome research.
Hanage says that even the two studies7,8 that analysed people in the United States with the same type of cancer identified only a partially overlapping set of microbes associated with positive outcomes. If researchers don’t work out the reason for these differences, they might not be able to interpret the outcomes of the trials, Hanage says.
Before starting clinical trials, the three groups should try to reproduce each other’s results and converge on a set of ‘beneficial’ microorganisms, Hanage argues. “Any of these bacteria could be a useful approach.” But inconsistencies might mean that the results are not reproducible.
It’s a concern common to microbiome research. “A lot of findings have proven to either not stand up or be considerably more complicated than they first appeared,” Hanage says. Standards such as those developed by the IHMS project should help, but scientists will be reluctant to take them on board, says Susan Erdman, a microbiologist and cancer biologist at the Massachusetts Institute of Technology in Cambridge. Doing so would come at the cost of innovation, she argues — it’s by experimenting in different settings that researchers make discoveries.
Wargo says that the community should standardize its approaches for collecting samples and doing analyses, as well as for validating studies in larger groups of patients. Since last year, her group has analysed stools from more than 500 people with skin cancer who had received different therapies. In parallel with the Paris-based team led by Zitvogel, the researchers are analysing patients treated with two combined immunotherapies to work out which gut bacteria mediate a response to that combination. Wargo hopes that the gut microbiome could eventually help to identify which patients will respond to which anticancer treatments. “Can we use it as a biomarker? It’s a provocative question,” she says.
In the short term, there will be a whole lot more sample collection. And this time around, it’s likely that fewer oncologists will raise an eyebrow, says Routy, who is now investigating how the gut microbiome boosts immunotherapy with his own group. In cancer therapy, “gut microbes have gone from ignored to super-popular organisms”, he says. Now, they’ll just have to live up to their reputation.
One-third of all protein-coding genes from bacterial genomes cannot be annotated with a function. Here, to investigate the functions of these genes, we present genome-wide mutant fitness data from 32 diverse bacteria across dozens of growth conditions. We identified mutant phenotypes for 11,779 protein-coding genes that had not been annotated with a specific function. Many genes could be associated with a specific condition because the gene affected fitness only in that condition, or with another gene in the same bacterium because they had similar mutant phenotypes. Of the poorly annotated genes, 2,316 had associations that have high confidence because they are conserved in other bacteria. By combining these conserved associations with comparative genomics, we identified putative DNA repair proteins; in addition, we propose specific functions for poorly annotated enzymes and transporters and for uncharacterized protein families. Our study demonstrates the scalability of microbial genetics and its utility for improving gene annotations.
As D.I.Y. Gene Editing Gains Popularity, ‘Someone Is Going to Get Hurt’
After researchers created a virus from mail-order DNA, geneticists sound the alarm about the genetic tinkering carried out in garages and living rooms.
Across the country, biohackers — hobbyists, amateur geneticists, students and enthusiasts — are practicing gene editing, concerning some bioterrorism experts.Ryan Christopher Jones for The New York Times
Across the country, biohackers — hobbyists, amateur geneticists, students and enthusiasts — are practicing gene editing, concerning some bioterrorism experts.Ryan Christopher Jones for The New York Times
WASHINGTON — As a teenager, Keoni Gandall already was operating a cutting-edge research laboratory in his bedroom in Huntington Beach, Calif. While his friends were buying video games, he acquired more than a dozen pieces of equipment — a transilluminator, a centrifuge, two thermocyclers — in pursuit of a hobby that once was the province of white-coated Ph.D.’s in institutional labs.
“I just wanted to clone DNA using my automated lab robot and feasibly make full genomes at home,” he said.
Mr. Gandall was far from alone. In the past few years, so-called biohackers across the country have taken gene editing into their own hands. As the equipment becomes cheaper and the expertise in gene-editing techniques, mostly Crispr-Cas9, more widely shared, citizen-scientists are attempting to re-engineer DNA in surprising ways.
Until now, the work has amounted to little more than D.I.Y. misfires. A year ago, a biohacker famously injected himself at a conference with modified DNA that he hoped would make him more muscular. (It did not.)
Earlier this year, at Body Hacking Con in Austin, Tex., a biotech executive injected himself with what he hoped would be a herpes treatment. (Verdict: No.) His company already had live-streamed a man injecting himself with a home-brewed treatment for H.I.V. (His viral load increased.)
In a recent interview, Mr. Gandall, now 18 and a research fellow at Stanford, said he only wants to ensure open access to gene-editing technology, believing future biotech discoveries may come from the least expected minds.
Keoni Gandall, in his laboratory at home in Palo Alto, Calif. He barely earned a high school diploma, he said, and was kicked out of a local science fair for reckless genetic engineering.Erin Brethauer for The New York Times
But he is quick to acknowledge that the do-it-yourself genetics revolution one day may go catastrophically wrong.
“Even I would tell you, the level of DNA synthesis regulation, it simply isn’t good enough,” Mr. Gandall said. “These regulations aren’t going to work when everything is decentralized — when everybody has a DNA synthesizer on their smartphone.”
The most pressing worry is that someone somewhere will use the spreading technology to create a bioweapon.
Already a research team at the University of Albertahas recreated from scratch an extinct relative of smallpox, horsepox, by stitching together fragments of mail-order DNA in just six months for about $100,000 — without a glance from law enforcement officials.
The team purchased overlapping DNA fragments from a commercial company. Once the researchers glued the full genome together and introduced it into cells infected by another type of poxvirus, the cells began to produce infectious particles.
To some experts, the experiment nullified a decades-long debate over whether to destroy the world’s two remaining smallpox remnants — at the Centers for Disease Control and Prevention in Atlanta and at a research center in Russia — since it proved that scientists who want to experiment with the virus can now create it themselves.
The study’s publication in the journal PLOS One included an in-depth description of the methods used and — most alarming to Gregory D. Koblentz, the director of the biodefense graduate program at George Mason University — a series of new tips and tricks for bypassing roadblocks.
Students at the Genspace lab in Brooklyn attending a “Biohacker Boot Camp” where participants can learn basic technical skills for homegrown genetics projects.Ryan Christopher Jones for The New York Times
“Sure, we’ve known this could be possible,” Dr. Koblentz said. “We also knew North Korea could someday build a thermonuclear weapon, but we’re still horrified when they actually do it.”
Experts urged the journal to cancel publication of the article, one calling it “unwise, unjustified, and dangerous.” Even before publication, a report from a World Health Organization meeting noted that the endeavor “did not require exceptional biochemical knowledge or skills, significant funds or significant time.”
But the study’s lead researcher, David Evans, a virologist at the University of Alberta, said he had alerted several Canadian government authorities to his poxvirus venture, and none had raised an objection.
Many experts agree that it would be very difficult for amateur biologists of any stripe to design a killer virus on their own. But as more hackers trade computer code for the genetic kind, and as their skills become increasingly sophisticated, health security experts fear that the potential for abuse may be growing.
“To unleash something deadly, that could really happen any day now — today,” said Dr. George Church, a researcher at Harvard and a leading synthetic biologist. “The pragmatic people would just engineer drug-resistant anthrax or highly transmissible influenza. Some recipes are online.”
“If they’re willing to inject themselves with hormones to make their muscles bigger, you can imagine they’d be willing to test more powerful things,” he added. “Anyone who does synthetic biology should be under surveillance, and anyone who does it without a license should be suspect.”
Authorities in the United States have been hesitant to undertake actions that could squelch innovation or impinge on intellectual property. The laws that cover biotechnology have not been significantly updated in decades, forcing regulators to rely on outdated frameworks to govern new technologies.
Work in the Genspace lab in Brooklyn. Similar facilities have opened worldwide.Ryan Christopher Jones for The New York Times
The cobbled-together regulatory system, with multiple agencies overseeing various types of research, has left gaps that will only widen as the technologies advance.
Academic researchers undergo strict scrutiny when they seek federal funding for “dual-use research of concern”: experiments that, in theory, could be used for good or ill. But more than half of the nation’s scientific research and development is funded by nongovernmental sources.
In 2013, a quest to create a glowing plant via genetic engineering drew almost half a million dollars through Kickstarter, the crowdfunding website.
“There really isn’t a national governance per se for those who are not federally or government funded,” said Dr. William So, a biological countermeasures specialist at the Federal Bureau of Investigation.
Instead, he said, the agency relies on biohackers themselves to sound the alarm regarding suspicious behavior.
“I do believe the F.B.I. is doing their best with what they have,” said Dr. Thomas V. Inglesby, director of the Johns Hopkins Center for Health Security in Baltimore.
“But if you really want to do this, there isn’t a whole lot stopping you.”
Underground Experimenters
The F.B.I. has befriended many white-hat biohacking labs, among them Genspace in Sunset Park, Brooklyn. Behind an inconspicuous steel door on a gritty, graffiti-lined street, biohackers-in-training — musicians, engineers, retirees — routinely gather for crash courses in genetic engineering.
Participants in “Biohacker Boot Camp” learn basic technical skills to use in homegrown genetics projects, like concocting algae that glows.
“The double helix is the most iconic image of the 20th century, perhaps rivaled only by the mushroom cloud,” the bootcamp’s leader, Michael Flanagan, said to a recent class.
Genspace’s entryway resembles a college dorm room, complete with sagging couch, microwave, mini-fridge. But the lab itself is palatial: two stories of white brick walls, industrial kitchen counters marked with dry-erase notes, shelves towering with glassware and reagents.
It’s a significant upgrade for Genspace. Daniel Grushkin, the co-founder, used to host bacterial experiments in his living room over pizza and beer.
The group later moved into arental for creatives— roboticists, organic fashion designers, miniature-cupcake makers — and constructed a makeshift lab using old patio screen doors. It was Mr. Grushkin who reached out to the F.B.I.
“People might be calling you because we are nonscientists doing science in a busted-up old building,” he recalled telling bureau agents. “But we aren’t a meth lab, and we aren’t bioterrorists.”
Mr. Grushkin has become a trailblazer in biohacking risk management, in part because he recognizes that letting neophytes manipulate live organisms is “less like a ‘hackerspace,’ more like a pet store.”
Josiah Zayner, a former NASA scientist turned biohacker, in Oakland, Calif.Erin Brethauer for The New York Times
He has posted community guidelines, forbidden infectious agents in the lab, and accepted a grant of almost $500,000 to design security practices for some four dozen similar labs across the country.
Most of them report not having heard so much as a greeting from the F.B.I. At many, the consequence for breaking safety guidelines is simply the loss of membership — leaving the perpetrator to experiment in isolation, but still among thousands of enthusiasts huddled online in Facebook groups, email listservs and Reddit pages.
Many find their inspiration in Josiah Zayner, a NASA scientist turned celebrity biohacker who straps a GoPro camera to his forehead and streams experiments on himself from his garage. He’s the man who tried to make his muscles bigger.
“This is just normal Scotch packing tape,” Mr. Zayner, chief executive of a biohacking start-up called The Odin, told hisYouTube audienceone summer night, muttering expletives as he stripped the top layer of skin from his forearm. “This is Day 1 of my experiment to genetically engineer myself.”
In an interview, Mr. Zayner conceded that among his biohacking followers, an accident — not a premeditated offense — was conceivable.
“I guess I can see why they don’t let the entire public have access to Ebola,” he said. “The risk is, if they’re working with Ebola and their house burns down, the Ebola could somehow get out.”
Even Mr. Zayner is apprehensive of the movement he helped begin; he plans to include live frogs in The Odin’s D.I.Y.-Crispr kits to encourage his followers to experiment on animals instead of themselves — or others.
“It’s going to be a matter of time before someone resynthesizes smallpox,” Mr. Gandall said.Erin Brethauer for The New York Times
“I have no doubt that someone is going to get hurt,” he said. “People are trying to one-up each other, and it’s moving faster than any one of us could have ever imagined — it’s almost uncontrollable. It’s scary.”
A Biological Arms Race
If nefarious biohackers were to create a biological weapon from scratch — a killer that would bounce from host to host to host, capable of reaching millions of people, unrestrained by time or distance — they would probably begin with some online shopping.
A site calledScience Exchange, for example, serves as a Craigslist for DNA, a commercial ecosystem connecting almost anyone with online access and a valid credit card to companies that sell cloned DNA fragments.
Mr. Gandall, the Stanford fellow, often buys such fragments — benign ones. But the workarounds for someone with ill intent, he said, might not be hard to figure out.
Biohackers will soon be able to forgo these companies altogether with an all-in-one desktop genome printer: a device much like an inkjet printer that employs the letters AGTC — genetic base pairs — instead of the color model CMYK.
A similar device already exists for institutional labs, called BioXp 3200, which sells for about $65,000. But at-home biohackers can start with DNA Playground from Amino Labs, an Easy Bake genetic oven that costs less than an iPad, or The Odin’s Crispr gene-editing kit for $159.
Tools like these may be threatening in the wrong hands, but they also helped Mr. Gandall start a promising career.
Mr. Gandall in a Stanford University bioengineering lab, preparing cells for long-term storage. He wants to ensure open access to gene-editing technology.Erin Brethauer for The New York Times
At age 11, he picked up a virology textbook at a church book fair. Before he was old enough for a driver’s permit, he was urging his mother to shuttle him to a research job at the University of California, Irvine.
He began dressing exclusively in red polo shirts to avoid the distraction of choosing outfits. He doodled through high school — correcting biology teachers — and was kicked out of a local science fair for what was deemed reckless home-brew genetic engineering.
Mr. Gandall barely earned a high-school diploma, he said, and was rebuffed by almost every college he applied to — but later gained a bioengineering position at Stanford University.
“Pretty ironic, after they rejected me as a student,” he said.
He moved to East Palo Alto — with 14 red polo shirts — into a house with three nonbiologists, who don’t much notice that DNA is cloned in the corner of his bedroom.
His mission at Stanford is to build a body of genetic material for public use. To his fellow biohackers, it’s a noble endeavor.
To biosecurity experts, it’s tossing ammunition into trigger-happy hands.
“There are really only two things that could wipe 30 million people off of the planet: a nuclear weapon, or a biological one,” said Lawrence O. Gostin, an adviser on pandemic influenza preparedness to the World Health Organization.
“Somehow, the U.S. government fears and prepares for the former, but not remotely for the latter. It baffles me.”
The genome of the flu virus has been fully sequenced in its native RNA form for the first time. Previously, all influenza genomes — as well as those of other viruses that store their genetic material as RNA — had been determined by copying the molecule into DNA. The native flu genome was generatedusing ‘nanopore’ sequencing technology, which reads RNA strands as they stream through a tiny molecular channel.
Virus de la gripe
RNA is chemically similar to its better known cousin, DNA. In cellular organisms, it serves as an intermediary between DNA-encoded genes and proteins, and performs other tasks in the cell. But many viruses — including those behindpolio,Ebolaand the common cold — store their genetic information as RNA, rather than DNA. Barnes, head of the CDC’s influenza-genomics team, says that no one had sequenced the virus’s RNA genome before because it seemed nearly impossible. Previous methods for sequencing native RNA strands involved degrading one chemical base, or letter, at a time, and these techniques have changed little since their invention in the late 1970s2. To compensate, nearly all ‘RNA sequencing’ instead uses a viral enzyme called reverse transcriptase, which copies RNA into sequencer-friendly DNA strands.
Tiny is mighty
Nanopores offer a simpler way of sequencing actual RNA molecules, such as viral genomes. The technology is based on applying electrical current across a nanoscalemolecular pore, and then measuring tell-tale current fluctuations as genetic material snakes through.
Fast track: nanopore sequencing identifies individual bases as a strand of DNA is passed through a pore.
In January, researchers at a leading developer of the technology, UK-based Oxford Nanopore Technologies, directly sequenced RNA using achocolate-bar-sizeddevicecalledthe MinION3. That effort looked at transcripts of messenger RNA, the family of RNA molecules that conveys information from DNA to build proteins.
Oxford Nanopore
Oxford Nanopore's MinION sequencer can read DNA fragments up to 10 kilobases long
Barnes’s team applied this method to the genome of influenza A, which is roughly 13,500 RNA letters long and composed of eight segments. Barnes says his team’s approach isn’t ready for prime time. It required a lot of flu virus, and to iron out inevitable sequencing errors, the raw data had to be processed many times over. But nanopore technology is advancing quickly, and Barnes hopes that with further improvements, direct sequencing of flu and other RNA viruses will become routine.
At the top of his and other scientists’ wish lists are methods for identifying chemical modifications to RNA.More than 100 have been identifiedso far, but researchers have little idea what most of them do, in large part because it has been impossible to study them systematically. The Oxford Nanopore team was able to detect two common RNA modifications, or tags. Birney, who is a paid consultant to the company, expects that the technology will be able to find many more once machine-learning algorithms are used to unpick the tags’ signatures.
Sequencing modified bases of RNA would be “a big deal” for the field, says Bryan Cullen, a virologist at Duke University in Durham, North Carolina. His team found last year4that a tag called m6A seems to alter the expression of influenza genes during infection in mice, which promotes viral replication. Current methods for detecting such modifications are time-consuming and expensive, he adds. Stacy Horner, co-director of Duke’s Center for RNA Biology, says that nanopore sequencing could also reveal hidden diversity in RNA viruses, which is lost with other methods that cobble together much shorter stretches of genetic material to generate a genome.
Although the methods aren’t yet perfect, Birney says, biologists are still excited about the possibility of soon being able to sequence entire viral genomes and other RNA molecules in their natural forms. “Suddenly, we’ve got the technology to do this. It’s kind of amazing.”